Archive Generated Run Files
Whenever possible, ENA provides access to two types of file for each run we present: the submitted file(s) and archive-generated file(s). Both are visible in the ENA Browser view for runs:
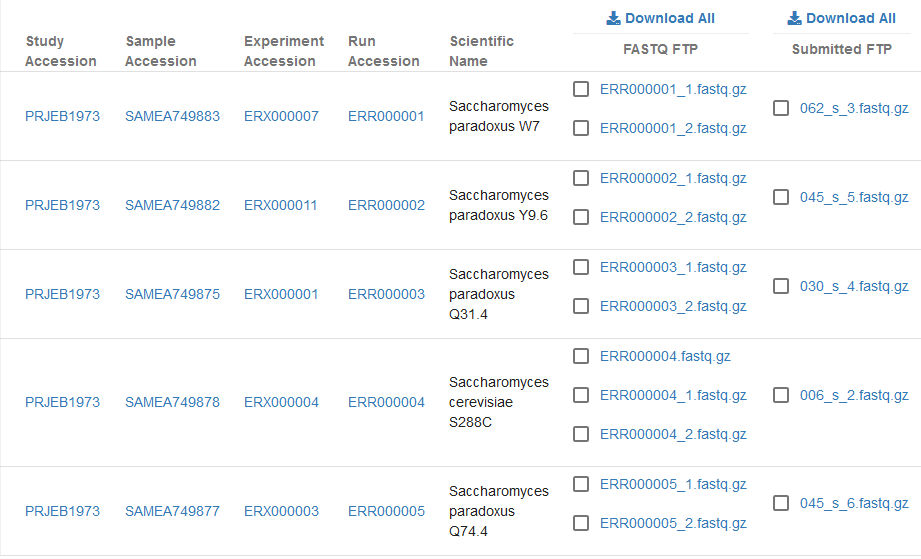
This page serves to briefly discuss the reason for this and the differences between the submitted and archive-generated files.
Submitted Files
The submitted files for any given run are copies of the files originally provided to us by the submitter. These files always undergo validation appropriate to their format, and are presented as-submitted with no automated curation. Formats are varied, and may be FASTQ but could also be others including BAM, FAST5, HDF5, etc.
Archive-Generated Files
Providing archive-generated FASTQs for runs is a means of bringing some consistency to the data we provide. By imposing a level of uniformity on these files, we can ensure users know what to expect of them and may incorporate them into pipelines with minimal friction.
Note that archive-generated FASTQ will not be available in the following uncommon scenarios:
BAM/CRAM files containing @PG:longranger
BAM/CRAM files containing @PG:cellranger
BAM/CRAM files containing CB:Z,CR:Z,CY:Z,RX:Z,QX:Z tags
Complete genomics native (data folder) submissions
PacBio native (HDF5) submissions
Many ONT native format submissions
Generated FASTQ Files
The number of files generated and their content varies depending on the nature of the submitted files
Number of
Application
Reads
|
FASTQ Files |
Description |
1 |
<run_accession>.fastq.gz
or
<run_accession>_1.fastq.gz
|
For experiments with single
application reads all reads will be
made available in one fastq file.
|
2 |
<run_accession>_1.fastq.gz
<run_accession>.fastq.gz
<run_accession>_2.fastq.gz
|
Paired experiments with two
application reads will be made
available in 1-3 FASTQ files. For a
paired experiment submitted with both
application reads the first reads
will be in <run accession>_1.fastq.gz
file, the second reads will be in
<run accession>_2.fastq.gz, and any
unpaired reads will be in <run
accession>.fastq.gz file. If files
from a paired experiment are
submitted and all reads are unpaired
then only a single file is created:
<run accession>.fastq.gz
|
> 2 |
<run_accession>_N.fastq.gz
|
For experiments with more than two
application reads (e.g. Complete
Genomics) one fastq file is created
for each application read, however,
no empty fastq files are created.
|
N/A |
<run_accession>_consensus.fastq.gz
|
ONT or PacBio consensus reads. |
N/A |
<run_accession>_subreads.fastq.gz
|
PacBio subreads. |
FASTQ File Format
@<run accession>.<spot index> [<spot name>][/<read index>]
<bases>
+
<phred qualities, ASCII encoded starting with '!' (33)>
Field |
Description |
<run accession> |
The run accession. A spot is identified uniquely by the combination
of the run accession and the spot index
|
<spot index> |
A positive integer assigned to the spots in the order in which they
appear in the run. A spot is identified uniquely by the combination of
the Run accession and the spot index.
|
<spot name> |
The spot name as it was provided by the submitter. In cases where the
read name is missing or was removed by the archive this field is not
present.
|
<read index> |
A positive integer assigned to the application reads in the order in
which they appear in the spot: /1 for first application read and /2 for
the second application read. In cases where the read name is missing or
was removed by the archive this field is not present.
|
Examples
Single layout:
@ERR000017.1 IL6_554:7:1:249:322
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
??????????????????????????????>>>>>>
Paired (first read):
@ERR005143.1 ID49_20708_20H04AAXX_R1:7:1:41:356/1
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh
Paired (second read):
@ERR005143.1 ID49_20708_20H04AAXX_R1:7:1:41:356/2
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh
Single layout without read names:
@ERR000017.1
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
??????????????????????????????>>>>>>
Paired without read names (first read):
@ERR005143.1
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh
Paired without read names (second read):
@ERR005143.1
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
+
hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh