Metagenome Submission Queries
Metagenomic studies involve assembling sequencing data sampled from an entire biome all the way down to the individual species that were living in that environment. As a result, there are often many queries regarding how these assemblies are submitted in order to make the quality of the assembly and original source of data as clear as possible.
The following image illustrates the stages of a metagenome assembly study and what is submittable to each of the metagenome assembly levels in ENA:
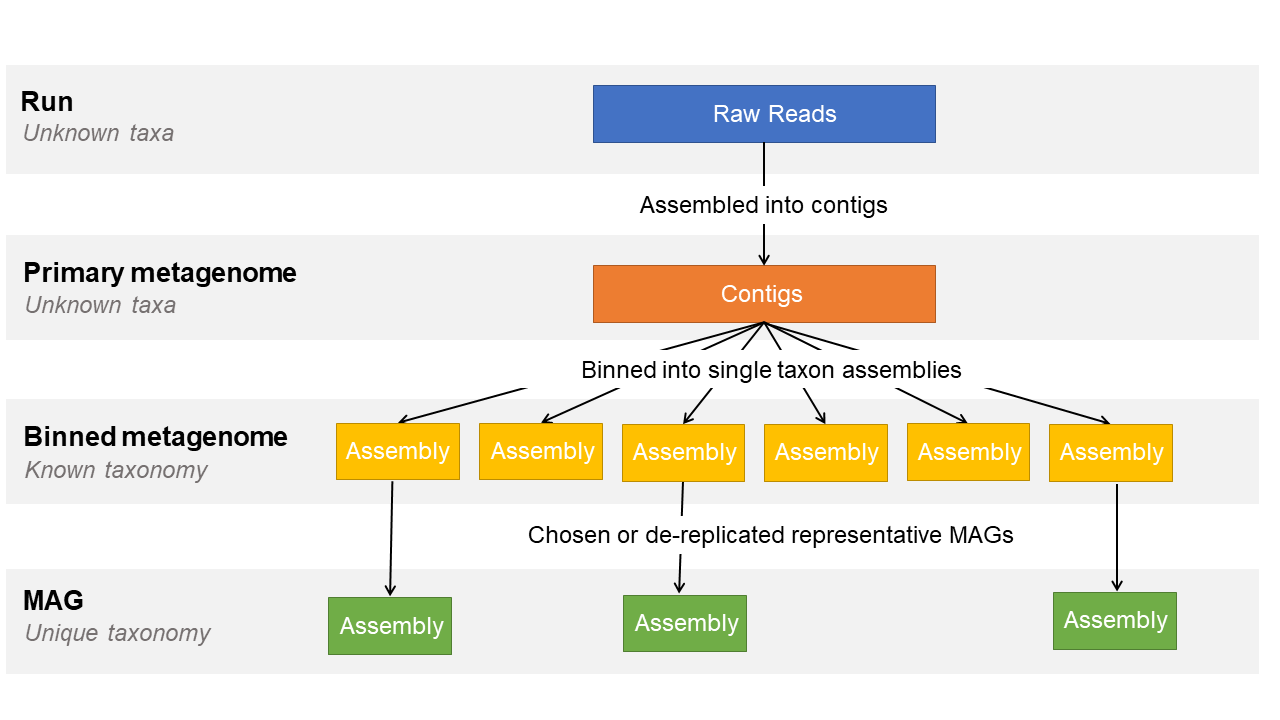
Please see here for more information on how to submit your metagenome assemblies to ENA.
What is defined as a MAG within ENA?
Within ENA, a MAG is described as a single-taxon assembly based on one or more binned metagenomes asserted to be a close representation to an actual individual genome (that could match an already existing isolate or represent a novel isolate).
There should only be one MAG submitted for each species within a biome. This can be determined using a de-replication step or by choosing the highest quality representative genome for each predicted species.
MAG assemblies are registered within ENA in the same domain as cultured isolate genome assemblies which means that these genome assemblies are searchable alongside cultured isolates and are used by the same downstream processes. As an environmental sample can contain many duplicate genomes of the same organism and as MAG assemblies are more prone to contamination, we request only the highest quality unique-taxon submissions are submitted as MAGs. This means only the highest quality sequences and most accurate annotated features are used within these downstream services to keep them as relevant and informative as possible.
How is the quality of a metagenomic assembly defined?
When binned and MAG samples are registered with ENA, three quality measures are recorded which together determine the overall quality of a metagenome derived assembly. These three measures follow the data standards defined by the Genomic Standards Consortium (GSC) in the 2018 publication here.
The three attributes in the sample checklists that contribute to the overall quality are as follows:
Assembly Quality - a written description of the quality of an assembly (one of a choice of 3 options).
Completeness Score - the ratio of observed single-copy marker genes to total single-copy marker genes in chosen marker gene set (%).
Contamination Score - the ratio of observed single-copy marker genes in ≥2 copies to total single-copy marker genes in chosen marker gene set (%).
It is essential you complete these fields accurately so that the overall quality of an assembly is searchable within ENA. If you wish to search for binned metagenomes or MAGs by overall quality, the thresholds for these standards are outlined below.
Finished Assembly
Any assembly where the assembly quality is defined as: “Single contiguous sequence without gaps or ambiguities with a consensus error rate equivalent to Q50 or better”.
High-quality draft
An assembly with the following criteria:
Attribute |
Value |
assembly quality |
Multiple fragments where gaps span repetitive regions. Presence of the 23S,
16S and 5S rRNA genes and at least 18 tRNAs.
|
completeness score |
>90% |
contamination score |
<5% |
Medium-quality draft
An assembly with the following criteria:
Attribute |
Value |
assembly quality |
Many fragments with little to no review of assembly other than reporting of
standard assembly statistics.
|
completeness score |
≥50% |
contamination score |
<10% |
Low-quality draft
An assembly with the following criteria:
Attribute |
Value |
assembly quality |
Many fragments with little to no review of assembly other than reporting of
standard assembly statistics.
|
completeness score |
<50% |
contamination score |
<10% |
It is useful to bear in mind these quality thresholds when assembling and submitting metagenomic assemblies.
How do I register samples for co-assemblies?
When registering co-assemblies, you should reference multiple samples (or reads) in the “sample derived from” field. These can be formatted as one of the following:
A comma separated list:
This should not contain spaces.
e.g. formatted as
ERSxxxxxx,ERSxxxxxx
or
ERRxxxxxx,ERRxxxxxx
A range:
This should be in the case where the assembly was derived from many samples and should be formatted with a ‘-‘ character between two accession without any spaces. The accession format should be consistent within the range and all accessions referenced within the range should have been used in that assembly.
e.g. formatted as
ERSxxxxxx-ERSxxxxxx
or
ERRxxxxxx-ERRxxxxxx
If you wish to submit a primary assembly which is co-assembled from raw reads, please inform our helpdesk.
How do I submit uncultured virus genomes (UViGs)?
The method used for submission of uncultured virus genomes depends on the methods used to identify these genomes.
If the virus genome was derived from a study where the entire biome of environmental data was sequenced together and then binned by taxonomy, then please submit with the same methods as those outlined in the metagenome assembly submission guidelines. This is with the exception of the use of the GSC MIMAGS checklist. For virus genomes, the GSC MIUVIGS checklist should be utilised for each virus assembly.
If the virus genome was derived from a study using single-cell amplification techniques, then please submit with the same methods as those outlined in the environmental single-cell amplified genome assembly submission guidelines. This is with the exception of the use of the GSC MISAGS checklist. For virus genomes, the GSC MIUVIGS checklist should be utilised for each virus assembly.
How do I submit metagenome assemblies without raw data or primary assemblies to point to?
It is recommended to submit all levels of metagenomic assembly where possible. However, there are exceptions where this can not be done. For example, if you have assembled bacteria from a metagenome derived from a human host, your raw data may be contaminated with human DNA which you do not have the permission to make publicly available.
In cases where it is not possible to provide raw data or a primary metagenome, environmental samples should still be registered. However, as the registered environmental samples do not have any data associated with them, they need to be manually released to become available to the public.
If you have not submitted raw reads or primary assemblies, sample release can be done in advance of your study release without the risk of any data files being prematurely released. However, if you do not wish to have your sample metadata publicly available before your study is released, this option is not suitable and it is recommended to make a note of the Study release date and release these samples during the same time of the study release.
To manually release your environmental samples, first you need to prepare a submission XML file containing all your environmental sample accessions in a block of ACTION tags.
An example of a submission XML for the release of three environmental samples is below:
<SUBMISSION>
<ACTIONS>
<ACTION>
<RELEASE target="ERS3334823"/>
</ACTION>
<ACTION>
<RELEASE target="ERS3334824"/>
</ACTION>
<ACTION>
<RELEASE target="ERS3334825"/>
</ACTION>
</ACTIONS>
</SUBMISSION>
These samples can then be released programmatically through the secure HTTPS protocol using a tool such as curl.
Below is an example of a environmental sample release command:
curl -u username:password -F "SUBMISSION=@submission.xml" "https://www.ebi.ac.uk/ena/submit/drop-box/submit/"
If your release is successful you should receive a receipt like the one below:
<RECEIPT receiptDate="2019-03-25T08:23:45.795Z" submissionFile="submission.xml" success="true">
<MESSAGES>
<INFO>sample accession "ERS3334823" is set to public status.</INFO>
<INFO>sample accession "ERS3334824" is set to public status.</INFO>
<INFO>sample accession "ERS3334825" is set to public status.</INFO>
<INFO>Submission has been committed.</INFO>
</MESSAGES>
<ACTIONS>RELEASE</ACTIONS>
<ACTIONS>RELEASE</ACTIONS>
<ACTIONS>RELEASE</ACTIONS>
</RECEIPT>