Submit Raw Reads Programmatically
Sequence read data is submitted using experiment and run XMLs.
An experiment object represents the library solution that is created from a sample and used in a sequencing experiment. The experiment object contains details about the sequencing platform and library protocols.
A run object represents a lane (or equivalent) on an sequencing machine and is used to attach sequence read data to experiments.
The experiment XML format is defined by SRA.experiment.xsd XML Schema, and the run XML format is defined by SRA.run.xsd XML Schema.
Object relationships
Both run and experiment are associated with other objects.
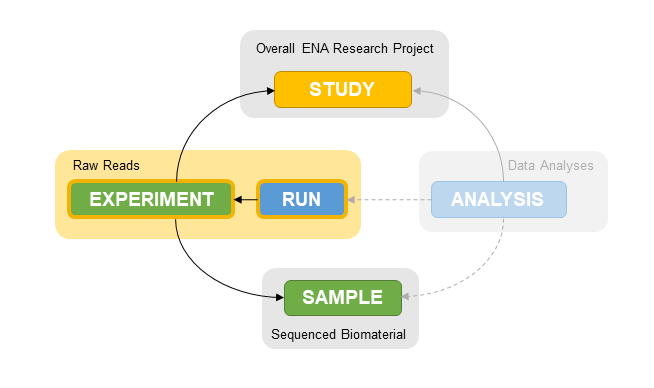
An experiment is part of a study. Studies are used to group together experiments to allow them to be cited together in a publication.
An experiment is assocated with a sample. It is common to have multiple libraries and sequencing experiments for a single sample. Experiments point to samples to allow sharing of sample information between multiple experiments.
To summarise object relationships:
One or more runs are part of an experiment.
One or more experiments are part of a study.
One or more experiments are associated with a sample.
It is common to pre-register samples ahead of submitting sequence reads. Note that samples and studies will be associated with each only after experiments have been submitted.
Run XML: part of experiment
A run points to the experiment it is part of using the <EXPERIMENT_REF> element. This can be done either by using an accession:
<EXPERIMENT_REF accession="ERX123456"/>
or a name within the submitter’s account:
<EXPERIMENT_REF refname="exp_mantis_religiosa"/>
Above, the refname refers to the submitter provided name (alias) of the experiment.
If the run is submitted at the same time as the experiment then the accession attribute can’t be used to refer to the experiment as the experiment accession has not been assigned yet. Had the experiment been submitted previously then the accession attribute could have been used.
When referring to experiments in other submission accounts, for example in a collaborators account, the accession attribute must be used. Names can be ambiguous between submission accounts as many other submitted objects could share the same name.
These principles with refname and accession attributes applies to all references between objects including experiment references to studies and samples.
Experiment XML: part of study
An experiment points to the study it is part of using the <STUDY_REF> element. In the example below this reference is made using an accession number:
<STUDY_REF accession="ERP123456"/>
Either an accession or a refname (alias) can be used in the reference. If you are using the accession attribute you can use both ERP and PRJ accessions when referring to studies.
Experiment XML: associated with sample
An experiment points to the sample it is associated with using the <SAMPLE_DESCRIPTOR> element. In the example below this reference is made using an accession number:
<SAMPLE_DESCRIPTOR accession="SRS462875"/>
Either an accession or a refname (alias) can be used in the reference. If you are using the accession attribute you can use both ERS and SAM accessions when referring to samples.
Metadata standards
The ENA is involved with minimum information standards for various project collaborations and sequencing fields. Even if you are not a member of one of these you can still use them as a guideline for increasing the quality of annotation of your experiment (and run) objects towards more interpretable and reproducible publications.
Supported data formats
Please see Read Data Formats.
Upload data files
Please see Data Upload.
You must have uploaded data files into your Webin upload area before you can submit them using a run XML. Once the run has been submitted the data files will be moved from the Webin upload area into the archive.
You can upload your data files to the root directory of your upload area or you can create subdirectories and upload your files there.
If the files are uploaded to the root directory then simply enter the file name in the Run XML when referring to it:
<FILE filename="mantis_religiosa_R1.fastq.gz" ... />
If the files are uploaded into a subdirectory (e.g. mantis_religiosa) then prefix the file name with the name of the subdirectory:
<FILE filename="mantis_religiosa/mantis_religiosa_R1.fastq.gz" ... />
Tip
Read names must not exceed a length of 256 characters. For more information on file format, see here.
Create the Run and Experiment XML
Run and Experiment XML: paired fastq
Below is an example of an Illumina HiSeq 2000 paired end reads being submitted in Fastq format.
The experiment points to a pre-registered sample and study using their accessions.
The run points to the experiment using the experiment’s alias.
Experiment XML:
<EXPERIMENT_SET>
<EXPERIMENT alias="exp_mantis_religiosa">
<TITLE>The 1KITE project: evolution of insects</TITLE>
<STUDY_REF accession="SRP017801"/>
<DESIGN>
<DESIGN_DESCRIPTION/>
<SAMPLE_DESCRIPTOR accession="SRS462875"/>
<LIBRARY_DESCRIPTOR>
<LIBRARY_NAME/>
<LIBRARY_STRATEGY>RNA-Seq</LIBRARY_STRATEGY>
<LIBRARY_SOURCE>TRANSCRIPTOMIC</LIBRARY_SOURCE>
<LIBRARY_SELECTION>cDNA</LIBRARY_SELECTION>
<LIBRARY_LAYOUT>
<PAIRED NOMINAL_LENGTH="250" NOMINAL_SDEV="30"/>
</LIBRARY_LAYOUT>
<LIBRARY_CONSTRUCTION_PROTOCOL>Messenger RNA (mRNA) was isolated using the Dynabeads mRNA Purification Kit (Invitrogen, Carlsbad Ca. USA) and then sheared using divalent cations at 72*C. These cleaved RNA fragments were transcribed into first-strand cDNA using II Reverse Transcriptase (Invitrogen, Carlsbad Ca. USA) and N6 primer (IDT). The second-strand cDNA was subsequently synthesized using RNase H (Invitrogen, Carlsbad Ca. USA) and DNA polymerase I (Invitrogen, Shanghai China). The double-stranded cDNA then underwent end-repair, a single `A? base addition, adapter ligati on, and size selection on anagarose gel (250 * 20 bp). At last, the product was indexed and PCR amplified to finalize the library prepration for the paired-end cDNA.</LIBRARY_CONSTRUCTION_PROTOCOL>
</LIBRARY_DESCRIPTOR>
</DESIGN>
<PLATFORM>
<ILLUMINA>
<INSTRUMENT_MODEL>Illumina HiSeq 2000</INSTRUMENT_MODEL>
</ILLUMINA>
</PLATFORM>
<EXPERIMENT_ATTRIBUTES>
<EXPERIMENT_ATTRIBUTE>
<TAG>library preparation date</TAG>
<VALUE>2010-08</VALUE>
</EXPERIMENT_ATTRIBUTE>
</EXPERIMENT_ATTRIBUTES>
</EXPERIMENT>
</EXPERIMENT_SET>
Run XML:
<RUN_SET>
<RUN alias="run_mantis_religiosa" center_name="">
<EXPERIMENT_REF refname="exp_mantis_religiosa"/>
<DATA_BLOCK>
<FILES>
<FILE filename="mantis_religiosa_R1.fastq.gz" filetype="fastq"
checksum_method="MD5" checksum="9b8932f85caa54e687eba62fca3edce2"/>
<FILE filename="mantis_religiosa_R2.fastq.gz" filetype="fastq"
checksum_method="MD5" checksum="183d6a24e0c3704e993bebe75bbbd989"/>
</FILES>
</DATA_BLOCK>
</RUN>
</RUN_SET>
You can submit several experiments and runs at the same time by using multiple <EXPERIMENT> and <RUN> blocks.
Experiment XML:
<EXPERIMENT_SET>
<EXPERIMENT alias="exp_01">
...
</EXPERIMENT>
...
<EXPERIMENT alias="exp_05">
...
</EXPERIMENT>
</EXPERIMENT_SET>
Run XML:
<RUN_SET>
<RUN alias="run_01">
<EXPERIMENT_REF refname="exp_01"/>
<DATA_BLOCK>
<FILES>
...
</FILES>
</DATA_BLOCK>
</RUN>
...
<RUN alias="run_05">
<EXPERIMENT_REF refname="exp_05"/>
<DATA_BLOCK>
<FILES>
...
</FILES>
</DATA_BLOCK>
</RUN>
</RUN_SET>
Change the XMLs by entering your own information and save it in two files, for example experiment.xml and run.xml.
Change the value of alias to be a unique name. You will need the unique name for example to refer to your experiment when adding run objects to it. An alias can be a short acronym but it should be meaningful and memorable in some way.
The <FILES> block in the run XML references the data files that are being submitted as part of the run.
To check the integrity of the file transfer an md5 checksum must be provided for each file. You can provide this by using the checksum_method=”MD5” and checksum attributes in the <FILE> element, or you can provide the MD5 checksum in file <file>.md5 in the same folder as the corresponding data file <file>.
Run XML: BAM
Below is an example of a RUN XML when reads are submitted in BAM format. Please note that filetype has been set to bam.
<RUN_SET>
<RUN alias="run_mantis_religiosa" center_name="">
<EXPERIMENT_REF refname="exp_mantis_religiosa"/>
<DATA_BLOCK>
<FILES>
<FILE filename="mantis_religiosa_R1.bam" filetype="bam"
checksum_method="MD5" checksum="9b8932f85caa54e687eba62fca3edce2"/>
</FILES>
</DATA_BLOCK>
</RUN>
</RUN_SET>
Run XML: CRAM
Below is an example of a RUN XML when reads are submitted in CRAM format. Please note that filetype has been set to cram.
<RUN_SET>
<RUN alias="run_mantis_religiosa" center_name="">
<EXPERIMENT_REF refname="exp_mantis_religiosa"/>
<DATA_BLOCK>
<FILES>
<FILE filename="mantis_religiosa_R1.cram" filetype="cram"
checksum_method="MD5" checksum="9b8932f85caa54e687eba62fca3edce2"/>
</FILES>
</DATA_BLOCK>
</RUN>
</RUN_SET>
Run XML: Multi-fastq
Below is an example of a RUN XML when multi-fastq data is submitted. Please note that read_type qualifiers are required for each file.
<DATA_BLOCK>
<FILES>
<FILE filename="single_cell_S1_L001_I1_001.fastq.gz"
filetype="fastq" checksum_method="MD5"
checksum="40d636c363e1a3a5232d3d1e2feb2c70">
<READ_TYPE>feature_barcode</READ_TYPE>
</FILE>
<FILE filename="single_cell_S1_L001_R1_001.fastq.gz"
filetype="fastq" checksum_method="MD5"
checksum="efdb826f2627c2ac449de5aaf2a61aab">
<READ_TYPE>paired</READ_TYPE>
<READ_TYPE>umi_barcode</READ_TYPE>
</FILE>
<FILE filename="single_cell_S1_L001_R2_001.fastq.gz"
filetype="fastq" checksum_method="MD5"
checksum="4b68b447d43eea5b5e75e23cfb4da82e">
<READ_TYPE>sample_barcode</READ_TYPE>
</FILE>
<FILE filename="single_cell_S1_L001_R3_001.fastq.gz"
filetype="fastq" checksum_method="MD5"
checksum="977a3fded781e6141359b47d7c6177a1">
<READ_TYPE>paired</READ_TYPE>
<READ_TYPE>cell_barcode</READ_TYPE>
</FILE>
</FILES>
</DATA_BLOCK>
Run XML: Oxford Nanopore
If you wish to submit your nanopore sequencing reads in their native FAST5 format, you must prepare an individual gzipped tar archive for each run. The run XML should look as follows:
<RUN_SET>
<RUN alias="run_mantis_religiosa" center_name="">
<EXPERIMENT_REF refname="exp_mantis_religiosa"/>
<DATA_BLOCK>
<FILES>
<FILE filename="mantis_religiosa.tar.gz" filetype="OxfordNanopore_native"
checksum_method="MD5" checksum="9b8932f85caa54e687eba62fca3edce2"/>
</FILES>
</DATA_BLOCK>
</RUN>
</RUN_SET>
Experiment XML: library information
The experiment contains a <LIBRARY_DESCRIPTOR> block in order to capture basic library information within the <LIBRARY_STRATEGY>, <LIBRARY_SOURCE> and <LIBRARY_SELECTION> elements. These are controlled value fields and the permitted values are listed in the SRA.experiment.xsd XML Schema.
...
<LIBRARY_DESCRIPTOR>
<LIBRARY_NAME/>
<LIBRARY_STRATEGY>RNA-Seq</LIBRARY_STRATEGY>
<LIBRARY_SOURCE>TRANSCRIPTOMIC</LIBRARY_SOURCE>
<LIBRARY_SELECTION>cDNA</LIBRARY_SELECTION>
<LIBRARY_LAYOUT>
<PAIRED NOMINAL_LENGTH="250" NOMINAL_SDEV="30"/>
</LIBRARY_LAYOUT>
<LIBRARY_CONSTRUCTION_PROTOCOL>Messenger RNA (mRNA) was isolated using the Dynabeads mRNA Purification Kit (Invitrogen, Carlsbad Ca. USA) and then sheared using divalent cations at 72*C. These cleaved RNA fragments were transcribed into first-strand cDNA using SuperScript?II Reverse Transcriptase (Invitrogen, Carlsbad Ca. USA) and N6 primer (IDT). The second-strand cDNA was subsequently synthesized using RNase H (Invitrogen, Carlsbad Ca. USA) and DNA polymerase I (Invitrogen, Shanghai China). The double-stranded cDNA then underwent end-repair, a single `A? base addition, adapter ligation, and size selection on anagarose gel (250 * 20 bp). At last, the product was indexed and PCR amplified to finalize the library prepration for the paired-end cDNA.
</LIBRARY_CONSTRUCTION_PROTOCOL>
</LIBRARY_DESCRIPTOR>
...
Experiment XML: paired ended reads
In experiment XML, the <LIBRARY_LAYOUT> element within the <LIBRARY_DESCRIPTOR> differentiates between single and paired ended reads. In the Experiment XML fragment below, we have described a paired ended experiment using the <PAIRED> element:
...
<LIBRARY_LAYOUT>
<PAIRED NOMINAL_LENGTH="250" NOMINAL_SDEV="30"/>
</LIBRARY_LAYOUT>
...
The attribute PAIRED NOMINAL_LENGTH is the average insert size. It is not the length of the reads. It is the average size of the fragments that are being sequenced.
The attribute NOMINAL_SDEV is the standard deviation of the fragment lengths. This attribute is not mandatory so you can omit it if you do not have this detail.
Experiment XML: single ended reads
In the Experiment XML fragment below, we have described a single ended experiment using the <SINGLE> element:
...
<LIBRARY_LAYOUT>
<SINGLE/>
</LIBRARY_LAYOUT>
...
Experiment and Run XML: attributes
Additional annotation can be provided for experiments and runs using attributes in the XMLs.
In experiment XML these attributes are captured using the EXPERIMENT_ATTRIBUTE block:
<EXPERIMENT_ATTRIBUTE>
<TAG>library preparation date</TAG>
<VALUE>2010-08</VALUE>
</EXPERIMENT_ATTRIBUTE>
In run XML these attributes are captured using the RUN_ATTRIBUTE block which is identical to the EXPERIMENT_ATTRIBUTE except for the name.
Annotations using attribute blocks should fit the type of object. For an experiment object you would expect annotations related to the library solution and for a run object you would expect annotations related to the NGS machine run.
Please refer to the metadata standards section in this module for information about recommended experiment and run attributes.
Create the Submission XML
To submit experiments or runs, you need an accompanying submission XML in a separate file. Let’s call this file submission.xml.
<SUBMISSION>
<ACTIONS>
<ACTION>
<ADD/>
</ACTION>
</ACTIONS>
</SUBMISSION>
The submission XML declares one or more Webin submission service actions. In this case the action is <ADD/> which is used to submit new objects.
The XMLs can be submitted programmatically, using CURL on command line or using the Webin Portal.
Submit the XMLs using CURL
CURL is a Linux/Unix command line program which you can use to send the experiment.xml, run.xml and submission.xml to the Webin submission service.
curl -u username:password -F "SUBMISSION=@submission.xml" -F "EXPERIMENT=@experiment.xml" -F "RUN=@run.xml" "https://wwwdev.ebi.ac.uk/ena/submit/drop-box/submit/"
Please provide your Webin submission account credentials using the username and password.
After running the command above a receipt XML is returned. It will look like the one below:
<?xml version="1.0" encoding="UTF-8"?>
<RECEIPT receiptDate="2017-08-11T15:07:36.746+01:00" submissionFile="sub.xml" success="true">
<EXPERIMENT accession="ERX2151578" alias="exp_mantis_religiosa" status="PRIVATE"/>
<RUN accession="ERR2094164" alias="run_mantis_religiosa" status="PRIVATE"/>
<SUBMISSION accession="ERA986371" alias="mantis_religiosa_submission"/>
<MESSAGES>
<INFO>This submission is a TEST submission and will be discarded within 24 hours</INFO>
</MESSAGES>
<ACTIONS>ADD</ACTIONS>
<ACTIONS>ADD</ACTIONS>
</RECEIPT>
Submit the XMLs using Webin Portal
XMLs can also be submitted interactively using the Webin Portal. Please refer to the Webin Portal document for an example how to submit a study using XML. Other types of XMLs can be submitted using the same approach.
The Receipt XML
To know if the submission was successful look in the first line of the <RECEIPT> block.
The attribute success will have value true or false. If the value is false then the submission did not succeed. In this case check the rest of the receipt for error messages and after making corrections, try the submission again.
If the success attribute is true then the submission was successful. The receipt will contain the accession numbers of the objects that you have submitted.
Test and production services
Note the message in the receipt:
<INFO>This submission is a TEST submission and will be discarded within 24 hours</INFO>
It is advisable to first test your submissions using the Webin test service where changes are not permanent and are erased every 24 hours.
Once you are happy with the result of the submission you can use the CURL command again but this time using the production service. Simply change the part in the URL from wwwdev.ebi.ac.uk to www.ebi.ac.uk:
curl -u username:password -F "SUBMISSION=@submission.xml" -F "EXPERIMENT=@experiment.xml" -F "RUN=@run.xml" "https://www.ebi.ac.uk/ena/submit/drop-box/submit/"
Similarly, if you are using the Webin Portal change the URL from wwwdev.ebi.ac.uk to www.ebi.ac.uk.