Submitting Genome Assemblies of Individuals or Cultured Isolates
Introduction
Genome assemblies can be submitted to the European Nucleotide Archive (ENA)
using the Webin command line submission interface with -context genome option.
Please contact our helpdesk if you intend to submit an assembly assembled from third party data.
Genome assembly submissions include plasmids, organelles, complete virus genomes, viral segments/replicons, bacteriophages, prokaryotic and eukaryotic genomes.
A genome assembly submission includes:
General assembly information
Study accession or unique name (alias)
Sample accession or unique name (alias)
Assembly name
Assembly type
Assembly program
Sequencing platform
Minimum gap length
Molecule type (genomic DNA, genomic RNA or viral cRNA)
Coverage
Free text description of the genome assembly (optional)
Contig sequences (if any)
Scaffold sequences (if any)
Chromosome sequences (if any)
Unlocalised sequences (if any)
Functional annotation (optional)
For assembly submission purposes, the term ‘chromosome’ should be understood to include organelles (e.g. mitochondria and chloroplasts), plasmids and viral segments.
The below image provides an outline of the workflow for submitting assemblies:
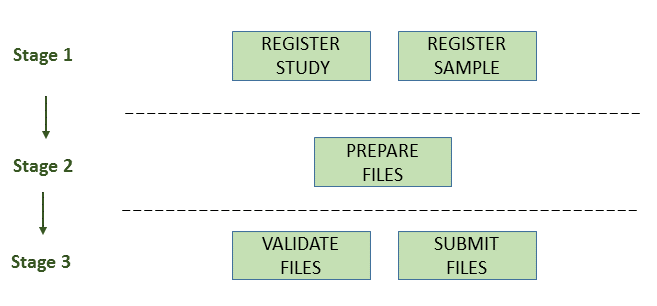
Stage 1: Pre-Register Study And Sample
Each submission must be associated with a pre-registered study and a sample.
Genome assemblies except primary metagenomes are uniquely associated with a study and a sample. When assemblies are updated they must be re-submitted with the same study and sample as in the original submission.
Tip
Check your sample has a submittable taxonomy. Your sample must have a binomial taxonomy. If the sample taxonomy is not binomial, then the genome assembly submission will fail validation.
It is also strongly recommended to submit the reads from which the assembly was assembled.
In order to reference the reads which were used to generate the assembly, please see the RUN_REF tag included in
the manifest file below.
Register locus tag prefixes
This is only required if you are submitting an annotated assembly. Otherwise, please proceed to Stage 2.
Locus tag prefixes should be registered with your study. See here for information on locus tags. Briefly, locus tag prefixes must:
Start with a letter
Be at least 3 characters long
Be upper case
Contain only alpha-numeric characters and no symbols such as -_*
You will need to wait 24 hours before proceeding further in the submission process to ensure these prefixes have been registered.
Stage 2: Prepare The Files
The set of files that are part of the submission are specified using a manifest file.
The manifest file is identified in the Webin-CLI command using the -manifest <filename> option.
Please note that the types of file which may be involved in an assembly submission are more comprehensively documented in our page Accepted Genome Assembly Data Formats.
The set of files required for submission of a genome assembly depends on the assembly level:
Contig Assembly
Consists of the following files:
1 manifest file
1 FASTA file OR 1 flat file
The default assumption is that an assembly is contig-level, thus if a submission comprises the sequences with no additional context, it is treated as a contig-level assembly
This assembly level only requires information on the sequences and annotation (if any). You will receive an error if only 1 sequence or more than 1,000,000 sequences are submitted. If you do not have a minimum of 2 contigs, then you will need to submit at a higher assembly level. If you have more than 1,000,000 contigs in your submission, please contact the helpdesk.
Seen an example contig-level assembly at: https://www.ebi.ac.uk/ena/browser/view/GCA_000003085
Scaffold Assembly
Consists of the following files:
This assembly level requires information on the sequences and annotation (if any). A scaffold is an assemblage of contigs separated by gaps of known length. These gaps define the scaffold, and so your submission must define them in one of two ways:
Explicit Gaps: Use an AGP file to describe how scaffolds are assembled from contigs in the sequence file
Implicit Gaps: Include gaps as runs of Ns in the sequence file and set the manifest file’s MINGAPLENGTH value to an appropriate integer value; each run of Ns which exceeds or matches this length will result in the creation of a scaffold
If an AGP file is included, it is not shown in our browser as part of the final assembly, but rather is consumed in the creation of the final sequence format. See an example scaffold-level assembly at: https://www.ebi.ac.uk/ena/browser/view/GCA_902705575
Chromosome Assembly
Consists of the following files:
This assembly level allows the submission of fully assembled replicons including chromosomes, organelles, plasmids, and viral segments. This requires information on the sequences and annotation (if any), and submission of a chromosome list file to indicate which sequences represent which ‘chromosomes’.
In addition to the chromosome sequences, you may include unlocalised and unplaced sequences. Unlocalised sequences (where the chromosome of the sequence is known but not the exact location) can be submitted with an additional unlocalised list file. Unplaced sequences (where the chromosome is entirely unknown) can be submitted the same as contigs; with no additional context. They will then be included as part of a contig accession set. Any sequences that are not used to assemble chromosomes are considered unplaced.
Note that all sequences should still be submitted in a single FASTA or flat file. Artificial constructs without biological evidence, such as artificial chromosomes consisting of unplaced contigs or scaffolds, are not permitted to be submitted.
See an example chromosome level assembly at: https://www.ebi.ac.uk/ena/browser/view/GCA_000237925
For this assembly level in particular, it is important to understand how sequence names are formatted so they can be consistent between files, otherwise the system will just register your submission at contig level.
Sequence Names
Sequences must have a unique name within the submission that is provided in the fasta, AGP or flat files. It is essential that the sequence names are unique and used consistently between files.
For example, the chromosome list file must refer to the chromosome sequences using the unique sequence names. Similarly, an AGP file must refer to scaffolds or contigs using the unique sequence names.
Manifest Files
The manifest file describes your assembly, including metadata and file names. It is a plain text file with two columns separated by a tab (or any whitespace characters):
Field name (first column): case insensitive field name
Field value (second column): field value
The following metadata fields are supported in the manifest file for genome context:
STUDY: Study accession - mandatory
SAMPLE: Sample accession - mandatory
ASSEMBLYNAME: Unique assembly name, user-provided - mandatory
ASSEMBLY_TYPE: ‘clone or isolate’ - mandatory
COVERAGE: The estimated depth of sequencing coverage - mandatory
PROGRAM: The assembly program - mandatory
PLATFORM: The sequencing platform, or comma-separated list of platforms - mandatory
MINGAPLENGTH: Minimum length of consecutive Ns to be considered a gap - optional
MOLECULETYPE: ‘genomic DNA’, ‘genomic RNA’ or ‘viral cRNA’ - optional
DESCRIPTION: Free text description of the genome assembly - optional
RUN_REF: Comma separated list of run accession(s) - optional
Please see further below for validation rules affecting some of these fields.
Various file name fields are supported in the manifest file. Note that all of these are optional, though of course at least one must be provided, and some may only be relevant in the presence of other file types. The available fields are as follows:
FASTA: sequences in fasta format
FLATFILE: sequences in EMBL-Bank flat file format
AGP: sequences in AGP format
CHROMOSOME_LIST: list of chromosomes
UNLOCALISED_LIST: list of unlocalised sequences
For example, the following manifest file represents a genome assembly consisting of contigs provided in one fasta file:
STUDY TODO
SAMPLE TODO
ASSEMBLYNAME TODO
ASSEMBLY_TYPE clone or isolate
COVERAGE TODO
PROGRAM TODO
PLATFORM TODO
MINGAPLENGTH TODO
MOLECULETYPE genomic DNA
FASTA genome.fasta.gz
Stage 3: Validate And Submit The Files
Files are validated, uploaded and submitted using the Webin command line submission interface (Webin-CLI). Please refer to the Webin command line submission interface documentation for full information about the submission process.
Brief examples of Webin-CLI commands follow.
The tool has -submit and -validate options which are mutually exclusive.
Full validation of your data and metadata is run regardless of which option you choose, but using just -validate
gives you the opportunity to check the current status of your assembly and information on any errors.
You are therefore encouraged to make use of Webin-CLI validation as much as you need to before you are ready to submit
for real.
First, run the Webin-CLI validation command, specifying your credentials and the path to your manifest file:
webin-cli -username Webin-XXXXX -password YYYYYYY -context genome -manifest manifest.txt -validate
Second, run the Webin-CLI submission command:
webin-cli -username Webin-XXXXX -password YYYYYYY -context genome -manifest manifest.txt -submit
In both cases, your prospective submission will be validated in full, and the result of this reported to you. A successful validation results in a simple success message, while a successful submission will further result in the assigned accession number (see below) being reported at your command line. Meanwhile, a failed validation will provide direction to a report file where you can find a list of error messages explaining the reason for the failure, which you can address before re-attempting.
For more information on how to install and use Webin-CLI, please refer to the Webin-CLI Submission page.
Assigned Accession Numbers
Once the genome assembly has been submitted an analysis (ERZxxxxxx) accession number is immediately assigned and returned to the submitter by the Webin command line submission interface (Webin-CLI).
ERZ accessions should not be used to reference the assembly in publications. The purpose of the ERZ accession number is for the submitter to be able to refer to their submission within the Webin submission service. For example, the submitter can retrieve the assigned genome assembly and sequence accessions from the Webin Portal or from the Webin reports service using the ERZ accession number. This accession should be used to refer to the assembly in any conversations with helpdesk staff.
For genome assemblies, long term stable accession numbers that can be used in publications are:
Study accession (PRJEBxxxxxx) assigned at time of study registration
Sample accession (SAMEAxxxxxx) assigned at time of study registration
Genome assembly accession (GCA_xxxxxx) assigned once the genome assembly has been fully processed by ENA and is released on agreement with GenCol. The GCA is stable between versions
Sequence accession(s) assigned once the genome assembly submission has been fully processed by ENA
Submitters can retrieve the genome and sequence accession numbers from the Webin Portal or from the Webin reports service. These accession numbers are also sent to the submitters by e-mail.
Validation Rules
Assembly submissions are subject to a great deal of validation before submission is allowed. Some key points are described here.
Sample And Study Validation
Sample and study (BioProject) pair must be unique for an assembly (except primary metagenomes)
Sample taxonomic classification must be species rank or below (or equivalent) within NCBI taxonomy.
Assembly Name Validation
Assembly names must:
match the pattern: ^[A-Za-z0-9][A-Za-z0-9 _#-.]*$
not be longer than 50 characters
not include the taxonomic name of the organism assembled
Chromosome Name Validation
Chromosome names must:
match the pattern: ^[A-Za-z0-9][A-Za-z0-9_#-.]*$
be shorter than 33 characters
be unique within an assembly
not contain any of the following as part of their name (case insensitive):
‘chr’
‘chrm’
‘chrom’
‘chromosome’
‘linkage group’
‘linkage-group’
‘linkage_group’
‘plasmid’
Sequence Validation
Sequences must:
have unique names within an assembly
be at least 20bp long
not have terminal Ns
consist of bases: ‘a’,’c’,’g’,’t’,’u’,’b’,’d’,’h’,’k’,’m’,’n’,’r’,’s’,’v’,’w’,’y’
Sequence Count Validation
Assembly submissions will typically not be allowed if the amount of sequences doesn’t fall within a required range. For example, contig-level assemblies must include more than 1 but less than 1,000,000 sequences. In specific cases, ENA may allow the submission of genome assemblies that are giving the following errors:
Since Webin 1.7.0:
Invalid number of sequences : XXX, Minimum number of sequences for CONTIG is: YYY
Invalid number of sequences : XXX, Minimum number of sequences for SCAFFOLD is: YYY
Invalid number of sequences : XXX, Minimum number of sequences for CHROMOSOME is: YYY
Invalid number of sequences : XXX, Maximum number of sequences for CONTIG is: YYY
Invalid number of sequences : XXX, Maximum number of sequences for SCAFFOLD is: YYY
Invalid number of sequences : XXX, Maximum number of sequences for CHROMOSOME is: YYY
This will be done at the discretion of the curation team when provided with valid reasoning, and can be requested through our helpdesk.
Note that their is no limit on the quantity of sequence data in bases, just the amount of sequences in total.
Assembly Updates
Assembly updates must: - use the same sample and study pair as was used in the initial assembly submission - not remove any chromosomes - use a unique ‘ASSEMBLY_NAME’ value in the manifest file
Automatic Fixes
Some fixes may be applied automatically, which users should be aware of. A few of these are documented below.
CDS Feature
Feature location is made 5’ partial if the /codon_start is 2 or 3.
Feature location is made 5’ partial if the /translation does not start with a start codon.
Feature location is made 3’ partial if the /translation does not end with a stop codon.
Feature location is made 3’ and 5’ partial if the location span is not a multiple of three.
Feature location has 3’ partiality removed if the /translation ends with a stop codon.
Feature is made /pseudo and the /translation is removed if the /translation contains internal stop codons.