Submitting Transcriptome Assemblies
Introduction
Transcriptome assemblies can be submitted to the European Nucleotide Archive (ENA) using the
Webin command line submission interface with -context transcriptome option.
A transcriptome assembly consists of:
General assembly information
Study accession or unique name (alias)
Sample accession or unique name (alias)
Assembly name
Assembly program
Sequencing platform
Free text description of the transcriptome assembly (optional)
Sequences
Functional annotation (optional)
The below image illustrates the stages of the transcriptome assembly submission process:
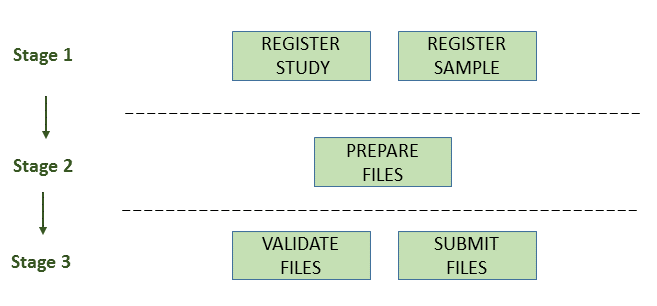
Stage 1: Pre-register study and sample
Each submission must be associated with a pre-registered study and a sample. For transcriptomic assemblies, raw reads must also be submitted to give context to the data.
Stage 2: Prepare the files
The set of files that are part of the submission are specified using a manifest file.
The manifest file is specified using the -manifest <filename> option.
A transcriptome assembly submission consists of the following files:
1 manifest file
1 FASTA file OR 1 flat file
Transcriptome assemblies should be of a high enough quality to fulfil the following criteria:
They must have at least 1x coverage by primary sequence at each base. Regions of a TSA record can be assembled from a single Expressed Sequence Tag (EST) or read so that coverage is only 1x.
Bases that are listed as ‘n’ should be less than 10%.
They should not have a stretch of more than 15 n’s in a row. If they are within 20 base pairs of the c- or n-terminus they should be removed.
No assemblies can be shorter than 200 base pairs.
Any vector sequence or primers should be removed.
Manifest file
The manifest file has two columns separated by a tab (or any whitespace characters):
Field name (first column): case insensitive field name
Field value (second column): field value
The following metadata fields are supported in the manifest file:
STUDY: Study accession or unique name (alias)
SAMPLE: Sample accession or unique name (alias)
ASSEMBLYNAME: The unique assembly name
ASSEMBLY_TYPE: ‘isolate’
PROGRAM: The assembly program
PLATFORM: The sequencing platform, or comma-separated list of platforms
DESCRIPTION: Free text description of the transcriptome assembly (optional)
RUN_REF: Comma separated list of run accession(s) (optional)
The following file name fields are supported in the manifest file:
FASTA: sequences in FASTA format
FLATFILE: sequences in EMBL-Bank flat file format
For example, the following manifest file represents a genome assembly consisting of transcripts provided in one FASTA file:
STUDY TODO
SAMPLE TODO
ASSEMBLYNAME TODO
ASSEMBLY_TYPE isolate
PROGRAM TODO
PLATFORM TODO
FASTA transcriptome.fasta.gz
Fasta file
Unannotated sequences should be submitted as a Fasta file.
The sequence name is extracted from the fasta header. For example the following header contains the name ‘contig1’:
>contig1
Flat file
Annotated sequences must be submitted using an EMBL-Bank flat file.
The sequence name is extracted from the AC * line and must be prefixed with a ‘_’. For example the following AC * line defines name ‘contig1’:
AC * _contig1
Stage 3: Validate and submit the files
Files are validated, uploaded and submitted using the Webin command line submission interface (Webin-CLI). Please refer to the Webin command line submission interface documentation for more information about the submission process.
Assigned accession numbers
Once the genome assembly has been submitted an analysis (ERZxxxxxx) accession number is immediately assigned and returned to the submitter by the Webin command line submission interface (Webin-CLI).
ERZ accessions should not be used to reference the assembly in publications. The purpose of the ERZ accession number is for the submitter to be able to refer to their submission within the Webin submission service. For example, the submitter can retrieve the assigned sequence accessions from the Webin Portal or from the Webin reports service using the ERZ accession number. Sequence accessions are sent to the user by email once the TSA has completed processing. The ERZ accession should be used to refer to the assembly in any conversations with helpdesk staff.
For transcriptome assemblies, long term stable accession numbers that can be used in publications are:
Study accession (PRJEBxxxxxx) assigned at time of study registration
Sample accession (SAMEAxxxxxx) assigned at time of study registration
Sequence accession(s) assigned once the genome assembly submission has been fully processed by ENA
See an example of a publicly available TSA at: https://www.ebi.ac.uk/ena/browser/view/GAAJ01000000
Validation rules
Sequence validation rules
Sequences must:
have unique names within an assembly
be at least 200bp long
not have terminal Ns
consist of bases: ‘a’,’c’,’g’,’t’,’u’,’b’,’d’,’h’,’k’,’m’,’n’,’r’,’s’,’v’,’w’,’y’