Submitting Binned Metagenome Assemblies
Introduction
Metagenome assemblies can be submitted to the European Nucleotide Archive (ENA) using the
Webin command line submission interface with -context genome.
Please contact our helpdesk if you intend to submit an assembly assembled from third party data.
A binned metagenome assembly submission encompasses anything from a set of contigs to a complete genome assembly from a metagenomic source that has been identified as a single-taxon set.
There is no limit to the number of bins that can be submitted as part of a metagenomic study as it is recognised that the number of bins produced can be very large. Please submit all derived bins at this assembly level and not as Metagenome-Assembled Genomes unless they meet the required criteria.
A binned metagenome assembly consists of:
General assembly information
Study accession or unique name (alias)
Binned Sample accession or unique name (alias)
Assembly program
Sequencing platform
Minimum gap length
Molecule type (genomic DNA, genomic RNA or viral cRNA)
Coverage
Free text description of the assembly (optional)
Contig sequences (if any)
Scaffold sequences (if any and submitting MAGs)
Chromosome sequences (if any and submitting MAGs)
The following picture illustrates the stages of the metagenome assembly submission process:
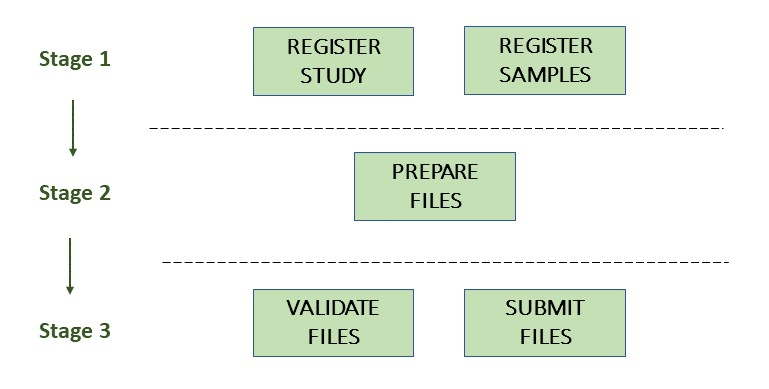
Stage 1: Pre-register study and metagenomic samples
Each submission must be associated with a pre-registered study and a binned sample.
If you have not done so already, please register a study.
It is recommended to submit any primary metagenomic assemblies and raw reads before submitting your binned metagenomes to help record your methods and make your data reproducible.
Registering binned samples
Each binned metagenome assembly submission must be associated with a binned sample. This is because a bin is not an assembly of all the data from the collected sample so linking to the environmental sample is misleading and causes incorrect taxonomy assignment. These binned samples represent the taxon sets derived from the environmental sample and hold all metadata related to the taxonomy of that subset as well as methods used to derive it.
These binned samples should be as specific in taxonomy as they can be and use the specific ENA binned metagenome checklist.
Please make sure these binned samples correctly reference the environmental sample that the bin was derived from. The environmental sample should be the same sample used to submit your raw reads and primary metagenomes.
This can be done from within the checklist using the mandatory “sample derived from” attribute. If the assembly was derived from multiple samples or runs you can list these with a comma separated list or range.
You should also reference the environmental sample in the description as one of the following:
“This sample represents a metagenomic bin from the metagenomic sample ERSXXXXX”
OR
“This sample represents a metagenomic bin from the metagenomic run ERRXXXXX”
The methods for submitting metagenomic samples follow the same process as any other sample submission. Follow the links for more information.
Stage 2: Prepare the files
The set of files that are part of the submission are specified using a manifest file.
The manifest file is specified using the -manifest <filename> option.
A binned metagenome assembly submission consists of the following files:
1 manifest file
1 fasta file
Manifest file
The manifest file has two columns separated by a tab (or any whitespace characters):
Field name (first column): case insensitive field name
Field value (second column): field value
The following metadata fields are supported in the manifest file:
STUDY: Study accession or unique name (alias)
SAMPLE: Binned sample accession or unique name (alias)
ASSEMBLYNAME: Unique assembly name
ASSEMBLY_TYPE: ‘binned metagenome’
COVERAGE: The estimated depth of sequencing coverage
PROGRAM: The assembly program
PLATFORM: The sequencing platform, or comma-separated list of platforms
MINGAPLENGTH: Minimum length of consecutive Ns to be considered a gap (optional)
MOLECULETYPE: ‘genomic DNA’, ‘genomic RNA’ or ‘viral cRNA’ (optional)
DESCRIPTION: Free text description of the genome assembly (optional)
RUN_REF: Comma separated list of run accession(s) (optional)
Please see further below for validation rules affecting some of these fields.
The following file name fields are supported in the manifest file:
FASTA: sequences in fasta format
For example, the following manifest file represents a binned metagenome assembly consisting of contigs provided in one fasta file:
STUDY TODO
SAMPLE TODO
RUN_REF TODO
ASSEMBLYNAME TODO
ASSEMBLY_TYPE TODO
COVERAGE TODO
PROGRAM TODO
PLATFORM TODO
MINGAPLENGTH TODO
MOLECULETYPE genomic DNA
FASTA binned_metagenome.fasta.gz
Stage 3: Validate and submit the files
Files are validated, uploaded and submitted using the Webin command line submission interface (Webin-CLI). Please refer to the Webin command line submission interface documentation for full information about the submission process.
Brief examples of Webin-CLI commands follow.
The tool has -submit and -validate options which are mutually exclusive.
Full validation of your data and metadata is run regardless of which option you choose, but using just -validate
gives you the opportunity to check the validation of your assembly and information on any errors.
You are therefore encouraged to make use of Webin-CLI validation as much as you need to before you are ready to submit
for real.
First, run the Webin-CLI validation command, specifying your credentials and the path to your manifest file:
webin-cli -username Webin-XXXXX -password YYYYYYY -context genome -manifest manifest.txt -validate
Second, run the Webin-CLI submission command:
webin-cli -username Webin-XXXXX -password YYYYYYY -context genome -manifest manifest.txt -validate
In both cases, your prospective submission will be validated in full, and the result of this reported to you. A successful validation results in a simple success message, while a successful submission will further result in the assigned accession number (see below) being reported at your command line. Meanwhile, a failed validation will provide direction to a report file where you can find a list of error messages explaining the reason for the failure, which you can address before re-attempting.
For more information on how to install and use Webin-CLI, please refer to the Webin-CLI Submission page.
Assigned accession numbers
Once the genome assembly has been submitted an analysis (ERZxxxxxx) accession number is immediately assigned and returned to the submitter by the Webin command line submission interface.
The purpose of the ERZ accession number is for the submitter to be able to refer to their submission within the Webin submission service and access their data in the browser.
For binned metagenome assemblies, long term stable accession numbers that can be used in publications are:
Study accession (PRJEBxxxxx) assigned at time of study registration.
Sample accession (SAMEAxxxxxx) assigned at time of sample registration.
See an example of a publicly available binned metagenome at: https://www.ebi.ac.uk/ena/browser/view/ERZ1100281
Validation rules
Assembly name validation
Assembly names must: - match the pattern: ^[A-Za-z0-9][A-Za-z0-9 _#-.]*$ - not be longer than 50 characters - not include the taxonomic name of the organism assembled
Sequence validation
Sequences must: - have unique names within an assembly - be at least 20bp long - not have terminal Ns - consist of bases: ‘a’,’c’,’g’,’t’,’u’,’b’,’d’,’h’,’k’,’m’,’n’,’r’,’s’,’v’,’w’,’y’